- Last edited on February 1, 2024
Introduction to Pharmacology
Primer
To prescribe medications, you must understand how drugs work and have a basic understanding of pharmacology. The two principle ideas in pharmacology are pharmacokinetics and pharmacodynamics:
- Pharmacokinetics: What the body (think kin- = body) does to the drug (through absorption, distribution, metabolism, and excretion).
- Pharmacodynamics: What the drug does to the body, the biochemical and physiological effects of drugs on the body. This includes the drug's mechanism of action, receptor binding, drug efficacy, drug potency, and toxicity.

PsychDB is an Amazon Associate and earns from qualifying purchases. Thank you for supporting our site!

Pharmacokinetics
There are four key principles of pharmacokinetics (also known as “ADME”):
- Absorption ‐ What is the route of drug delivery and where is it absorbed?
- Distribution ‐ Where does the drug go (does it stay in the blood or go into body fat or tissues?), where does it need to go (is there a target organ?), and what are the implications?
- Metabolism ‐ Where is the drug broken down?
- Excretion/Elimination – How is the drug eliminated?
Absorption
Before a drug can begin to exert any effect on the body it has to be absorbed into the body systems. This absorption process can be affected by many things but the main factor relating to absorption is the route of administration.
Absorption: Routes of drug administration
| Routes | Advantages | Disadvantages |
|---|---|---|
| Oral (PO) | Convenient | GI upset |
| Sublingual (SL) | Avoid first-pass metabolism and gastric acid (therefore GI upset) | Few preparations |
| Rectal (PR) | Avoid first-pass metabolism and gastric acid (therefore GI upset) | Few preparations, patient comfort |
| Intravenous (IV) | Rapid action, immediate availability | Increased levels to cardiac tissue, risk of sepsis/embolism if not sterile |
| Intramuscular (IM) | Rapid absorption | Pain, tissue damage risk |
| Subcutaneous (SC) | Slower absorption | Variable absorption |
Oral Absorption and First-Pass Metabolism
When a drug is consumed orally, it goes through a process called first-pass metabolism in the liver. First-pass metabolism reduces the bio-availability (fraction of drug absorbed into the systemic circulation) of the drugThe drug goes from the mouth → GI tract → portal vein → liver (first-pass metabolism happens here) → systemic circulation → drug target or receptor. When medications are given intramuscularly, sublingually, or rectally, this bypasses first-pass metabolism.
Distribution
Once a drug is administered and absorbed, it has to be distributed to their site of action. Some drugs go into a specific organ (e.g. - iodine into the thyroid gland). Other drugs are lipophilic and highly absorbed into fatty tissue (e.g. - most antidepressants and antipsychotics). Finally, other drugs (e.g. - lithium) are mostly distributed in body water (plasma, interstitial fluid and intracellular fluid).
Volume of Distribution (Vd) is one way to measure and quantify this drug distribution. Volume of distribution is affected by two main factors: fat stores and body water (total and extracellular). Other factors like protein binding and passage through barriers also affect distribution.
What is Volume of Distribution?
Volume of Distribution is a proportion factor that gives you the relationship between the amount of drug in the body (mg) to the plasma concentration of drug measured in a biological fluid (mg/L).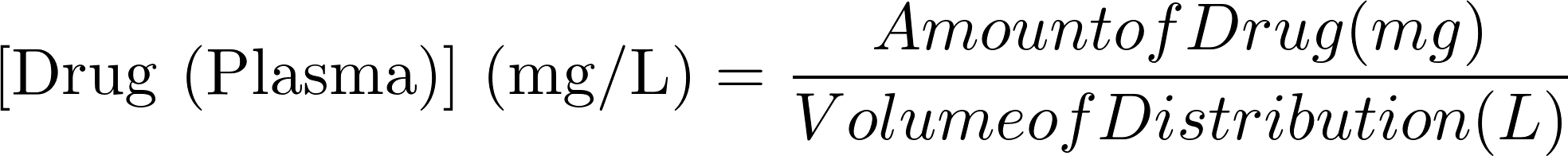
A larger volume of distribution means the drug is more likely to be in the tissues of the body, while a smaller volume of distribution means the drug is confined to the circulatory system.
Interpreting Volume of Distribution
| Volume of Distribution (Vd) | Distribution of Drug | Types of Drug |
|---|---|---|
| Low (< 4 )L | Mostly confined to the plasma component of blood (intravascular) | Large/charged molecules; plasma protein bound |
| Medium (4 to 7 L) | Distributed throughout the blood (both plasma and red blood cells), and extracellular fluid (ECF) | Small hydrophilic (water-loving) molecules |
| High (> 42 L*) | Distributed to all tissues in the body (including fatty tissue) | Small lipophilic (fat-loving) molecules, especially if bound to tissue protein |
| Very high (> 5000 L) | Mostly in the tissue, and very little of it is in the blood or plasma | Highly lipophilic molecules |
Two Main Factors That Affect Volume of Distribution (Vd)
| Population | Effect on Vd | Outcome | Clinical Implications | |
|---|---|---|---|---|
| Lower proportion of body fat | Children | ↓ Vd | For lipophilic drugs (e.g. - antidepressants and antipsychotics), a smaller Vd results in higher plasma drug concentrations | No significant effect, possibly due to increased clearance and elimination in children and adolescents |
| Higher proportion of body fat | Elderly | ↑ Vd | For lipophilic drugs (e.g. - antidepressants and antipsychotics), a larger Vd results in more storage of the drug in fatty tissue | Lipophilic drugs can accumulate in fat, remain in the body longer, and thus have a longer half-life. With repeated dosing, drugs accumulate in fat, and may be erratically released. Thus, drugs should be prescribed in a 'start low and go slow' method |
| Higher proportion of body water | Children | ↑ Vd | For drugs primarily distributed in body water (e.g. - lithium), a larger Vd results in lower plasma drug concentration | - |
| Lower proportion of body water | Elderly | ↓ Vd | For drugs primarily distributed in body water (e.g. - lithium), a smaller Vd results in higher plasma drug concentration | Lower doses of lithium may be effective in the elderly |
Other Factors Affecting Distribution
| Factors | Description |
|---|---|
| Plasma Protein Binding | Plasma proteins like albumin bind drug molecules. The degree of binding varies widely among drugs. Drugs bound to plasma proteins are pharmacologically inert – only the free drug is active. Some drugs do not bind at all (e.g. - caffeine) while others are highly bound (e.g. - warfarin which is 99% bound). Some drugs can displace others from their binding sites on the plasma proteins (e.g. - phenylbutazone can displace warfarin from plasma proteins). |
| Passage through Barriers | The two main barriers in the body are the placenta and the blood-brain barrier (BBB). For example, drugs must be highly lipid soluble in order to pass the blood-brain barrier to exert their action. This is why most psychotropic drugs (e.g. - antidepressants and antipsychotics) are highly lipid-soluble. |
Metabolism
Metabolism is the breaking down of a drug so it can be eliminated from the body. Most psychotropic drugs undergo extensive biotransformation in the liver in two phases (Phase I and Phase II):
- Phase I reactions (Cytochrome P450 System):
- Hydroxylation, reduction, and hydrolysis, catalyzed by hepatic microsomal enzymes
- Older adults typically lose or have decreased Phase I metabolism
- Children and adolescents typically have the highest (peak) Phase I metabolism
- The purpose of CYP metabolism is to make fat-soluble molecules more water-soluble so that they can be eliminated via the kidneys
- Phase II reactions (conjugation reactions, glucuronidation, acetylation, and sulfation):
- Phase II reactions couple a drug or a metabolite to an endogenous conjugating molecule such as glucuronic acid, sulfuric acid, acetic acid, or glutathione.
- Conjugation by glucuronic acid (“glucuronidation”) and occurs in almost any organ, but again primarily in the liver.
- Some drugs will undergo only Phase II reactions, and not Phase I reactions (e.g. - lorazepam, lamotrigine).
- Phase II reactions are metabolized at the same rate in children (>3 years) as in adults and older adults.
Both phase I and phase II reactions are susceptible to inhibition or induction by other drugs. For example:
- Phase I: fluoxetine increases atomoxetine, risperidone, and aripiprazole levels through CYP2D6 inhibition.
- Phase II: valproic acid increases lamotrigine levels through inhibition of glucuronidation.
Not All Drugs Are Metabolized!
Not all drugs are metabolized. Examples include lithium and gabapentin.Oral Absorption and First-Pass Metabolism
When a drug is consumed orally, it goes through a process called first-pass metabolism in the liver. First-pass metabolism reduces the bio-availability (fraction of drug absorbed into the systemic circulation) of the drugThe drug goes from the mouth → GI tract → portal vein → liver (first-pass metabolism happens here) → systemic circulation → drug target or receptor. When medications are given intramuscularly, sublingually, or rectally, this bypasses first-pass metabolism.
What is Glucuronidation?
Conjugation with glucuronic acid (glucuronidation) is the major route for the biotransformation and elimination of carboxylic acid-containing drugs. Glucuronidation is when a glucuronic acid is added to a substrate from UDP-glucuronic acid via a UDP glucuronosyltransferase.Excretion/Elimination
Elimination refers to the removal of a drug from the body. The kidney is the most important organ for drug elimination. Clearance refers to the efficiency of drug removal (i.e. - the theoretical volume of blood that is totally cleared of drug per unit time).
Half-life
The half-life is time required to reduce the plasma concentration of a drug to one half of its initial value. A drug with a half-life of 24 hours will reach steady state in approximately 5 days; similarly, one would need to stop taking this drug for 5 days in for it to be fully “out of the system.”
Steady State
Steady state occurs when the rate of drug absorption into the blood equals the rate of elimination from the body. The time to reach steady state concentrations depends on the half-life of the drug. Drugs with first-order elimination kinetics require 5 half-lives to reach steady state with repeated dosing. Similarly, 5 half-lives is needed for the drug to be completely eliminated from the body once the last dose is taken (see figure 2).
 Fig. 2: Steady state example with a 100 mg drug with a half life of 24 hours
Fig. 2: Steady state example with a 100 mg drug with a half life of 24 hours
Pharmacodynamics
Pharmacodynamics is the effect of the drug on the body. There are 7 main drug actions that can occur on the body:
- Stimulating action through direct receptor agonism and downstream effects (e.g. - dopamine agonists for Parkinson's)
- Depressing action through direct receptor agonism and downstream effects (e.g. - inverse agonist)
- Blocking/antagonizing action where the drug binds the receptor but does not activate it (e.g. - silent antagonists)
- Stabilizing action where the drug seems to act neither as a stimulant or as a depressant (e.g. - some drugs possess receptor activity that allows them to stabilize general receptor activation, such as buprenorphine in opioid use disorder, or aripiprazole in schizophrenia)
- Exchanging/replacing substances or accumulating them to form a reserve (e.g. - glycogen storage)
- Direct beneficial chemical reaction (e.g. - free radical scavenging)
- Direct harmful chemical reaction (e.g. - damage or destruction of the cells through induced toxic or lethal damage, also called cytotoxicity or irritation)

Affinity
Affinity is how avidly a drug binds its receptor or how the chemical forces that cause a substance to bind its receptor. The higher affinity that a drug (also known as a ligand) has for a receptor the “cleaner” the drug. Low affinity drugs are called “dirty” drugs because they bind to many receptor types, including those that are not the intended targets.
Potency
Potency is a measure of the quantity of drug needed to produce a maximal effect. That is, how much of a drug do you need (e.g. - 2mg vs. 200mg?) to give to achieve a desired effect.
Efficacy
Confusion Alert!
The term efficacy is also used in epidemiology, but describes a completely different concept!Efficacy is the maximum effect that a drug can produce regardless of dose.[1]